| 23. Protein assembly & tunover |
|
|
|
|
|
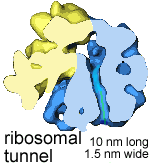
New amino acids are added to the polypeptide's C-terminus, while the N-terminus moves through a tunnel within the ribosome; about 15 amino acids are added per second. The ribosomal tunnel is approximately 10 nm long and 1.5 nm in diameter; it is narrow enough to block the folding of the newly synthesized polypeptide chain. |
 |
|
Translation, like most all processes that occur within the cell, is a stochastic process, meaning that it is based on random collisions between molecules. In the specific case of translation, the association of the mRNA with the ribosomal components occurs stochastically; similarly, the addition of a new amino acid depends on the collision of the appropriate amino acid-charged, tRNAs with the RNA-ribosome complex. Since there are many "inappropriate" amino-acid charged tRNAs in the cytoplasm, the ribosomal complex must be able to productively bind only the tRNA the mRNA specifies, that is the tRNA with the right anticodon. This enables its attached amino acid can interact productively to add the amino acid to the growing polypeptide chain. |
 |
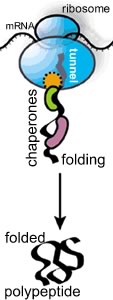
In most illustrations of polypeptide synthesis, you rarely see this fact illustrated. As the polypeptide emerges from the tunnel, it encounters the crowded cytoplasmic environment; at the same time it begins to fold. As it folds, the polypeptide needs to avoid non-specific interactions with other cellular components. If it is part of a multisubunit protein, it must "find" its partner polypeptides, again, a stochastic process. |
|
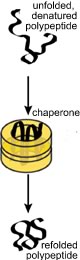
If the polypeptide does not fold correctly, it will not function correctly and may damage the cell. A number of degenerative neurological disorders are due, at least in part, to the accumulation of misfolded polypeptides (see below). A number of enzymes, known generically as chaperones, associate with the nascent polypeptide as it emerges from the ribosome. Chaperones facilitate the proper folding of the polypeptide, primarily by suppressing or unfolding 'incorrect' structures (perhaps you can guess how a chaperone "knows" something bad is going on). Chaperones bind to, and stabilize polypeptides prior to their final assembly into multisubunit complexes, the final active protein |
 |
|
|
|
|
One class of chaperones are known as heat shock proteins. These proteins recognize unfolded polypeptides - protein unfolding occurs with increasing frequency at higher temperatures. Heat-shock chaperones couple ATP hydrolysis reactions with polypeptide unfolding reactions to unfold misfolded polypeptides. They then release the unfolded polypeptide or protein, giving it another chance to refold correctly, on its own. How do chaperones recognize unfolded or abnormally folded proteins? They look for hydrophobic regions on their surface. Normal soluble proteins do not have such regions and the presence of such domains can lead to non-specific protein-protein aggregates. |
|
|
|
Protein degradation: If a protein does not fold correctly, it is generally degraded by enzymes known as proteases, enzymes that cleave proteins. This involves forming a complex with the protease. Proteases cleave peptide bonds via a hydrolysis reaction. The hydrolysis of a protein is a thermodynamically favorable reaction. It is the case that proteins differ in the probability that they will form a productive complex with a protease. In this case, a productive interaction means that the protein will be degraded. The process of degradation is known as turnover, in part because the amino acids released can be reused by the cell. The end result of the processes of translation (protein synthesis), folding, and degradation is that each protein in a cell has a half-life. If we had a population of molecules, the half-life is the time at which we would expect 50% of the those molecules present at time = 0 to have been degraded through interactions with proteases. There are a number of mechanisms involved in the turnover of proteins. |
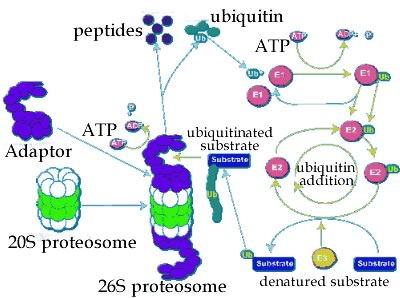 |
A common one involves a macromolecular machine, the proteosome. The proteosome is a multienzyme complex that degrades polypeptides. The proteases can be divided into two generic classes. Those that hydrolyze polypeptides from one end or the other, to release one or twp amino acid at a time, are known as exoproteases. Please note (in the video below), the unrealistic components. |
|
|
|
Proteases that cleave a polypeptide chain internally are known as endoproteases - they generate two polypeptides. Whether a protein is degraded depends upon how it interacts with other molecules, particularly chaperones and proteosomes, and other proteases [a mathematical analysis of protein (and RNA) half-life is beyond the scope of this course, but you can read about it here and here if you wish.] |
|
|
|
Diseases of folding and misfolding: If the functional protein is in its native state, a dysfunctional misfolded protein is said to be denatured. It does not take much of a perturbation to unfold or denature most proteins. In fact, under normal conditions, proteins often become partially denatured. A number of diseases have been associated with protein misfolding. |
|
Kuru was among the first of these to be identified. Beginning in the 1950s, D. Carleton Gadjusek studied a neurological disorder common among the Fore people of New Guinea. The symptoms of kuru, which means "trembling with fear", are similar to those of scrapie, a disease of sheep, and variant Creutzfeld-Jakob disease (vCJD) in humans. |
 |
|
Among the Fore people, kuru was linked to the ritual eating of the dead. Since this practice has ended, the disease has disappeared. |
 |
Prions and protein folding: The cause of kuru, scrapie and vCJD appears to be the presence of an abnormal form of a normal protein, known as a prion. We can think of prions as an anti-chaperone. The idea of proteins as infectious agents was championed by Stan Prusiner. |
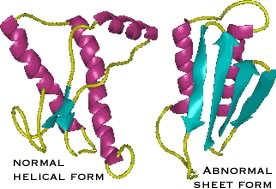
| The protein responsible for kuru and scrapie is known as PrPc. It normally exists in a largely α-helical form. There is a second abnormal form of the protein, PrPsc for scrapie; it is composed primarily of β-sheet. |
|
The two polypeptides have the same primary sequence. PrPsc acts as an anti-chaperone, catalyzing the transformation of PrPc into PrPsc. Once initiated, this leads to a chain reaction and PrPsc accumulation. As it accumulates, PrPsc assembles into rod-shaped aggregates that appear to damage cells. |
 |
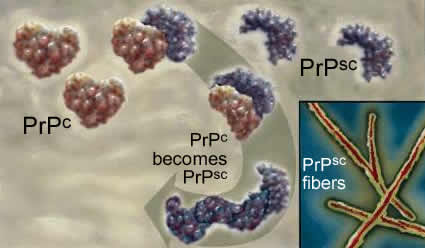 |
This process occurs within the cells of the central nervous system, and leads to severe neurological defects. There is no natural defense, since the protein responsible is a normal protein. |
|
Disease transmission: When the Fore ate the brains of their beloved ancestors, they inadvertently introduced the PrPsc protein into their bodies. Genetic studies indicate that early humans evolved resistance to prion diseases, suggesting that cannibalism might have been an important selective factor during human evolution. |
 |
|
Since cannibalism is not nearly as common today, how does anyone get such diseases in the modern world? There are rare cases of iatrogenic transmission, that is, where the disease is caused by faulty medical practice, for example through the use of contaminated surgical instruments or diseased tissue for transplantation. Where did these people get the disease? Since the disease is caused by the formation of PrPsc, any event that leads to PrPsc formation could cause the disease. Normally, the formation of PrPsc from PrPc is very rare. We all have PrPc but very few of us spontaneously develop kuru-like symptoms. There are, however, mutations in the gene that encodes PrPc that greatly enhance the frequency of the PrPc → PrPsc conversion. |
|
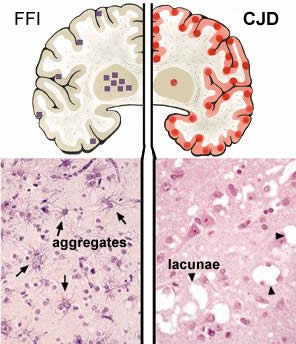
Such mutations may be inherited (genetic) or may occur during the life of an organism (sporadic). The schematic of the brain on the left is from FFI is due to the inheritance of a mutation in the PRNP gene; this mutation changes the normal aspartic acid 178 to an asparagine. The squares mark brain regions affected; the lower section reveals the presence of aberrant protein aggregates in these regions. When combined with a second mutation in the PRNP gene at position 129, the FFI mutation leads to Creutzfeld-Jacob disease (CJD). |
 |
|
The circles in the right hand brain schematic indicate regions affected in CJD. There are holes or lacunae in the tissue. Both FFI and CJD are late onset diseases. Symptoms first appear ~ 50 years of age. |
|
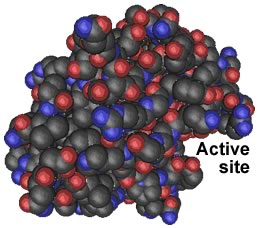
So why do PrPsc aggregates accumulate? To cut a peptide bond, a protease must position the target peptide bond within its catalytic active site.If the target protein's peptide bonds do not fit into the active site, they cannot be cut. Because of their structure, PrPsc aggregates are highly resistant to proteolysis. They gradually accumulate over many years, a fact that may explain the late onset of PrP-based diseases. |
 |
|
for the pre-medical students along you.
|
|
|
|
Questions to answer |
|
|
Questions to ponder
|
|
|
|
| replace with revised beSocratic activity |
|
|
|
|
Tweet
revised
10-May-2014
|
|